电子科技大学王晓霆教授团队在《化学理论与计算杂志》上发表面向较大分子体系的几何优化量子算法
近日,电子科技大学基础与前沿研究院王晓霆教授和北京大学袁骁团队在Journal of Chemical Theory and Computation发表题为“Large-Scale Efficient Molecule Geometry Optimization with Hybrid Quantum–Classical Computing”的研究论文。基础院博士研究生郝亚杰为论文第一作者,王晓霆教授和北京大学袁骁教授为共同通讯作者。电子科技大学基础与前沿研究院为论文第一单位。
分子几何结构是理解化学反应、材料性质和药物分子功能的基础。键长、键角和构型的微小变化,往往会显著影响分子的稳定性、反应活性以及与其他分子的相互作用。因此,如何高精度、高效率地预测复杂分子的平衡几何结构,是量子化学、药物设计和材料计算中的核心问题之一。
传统高精度电子结构方法在处理强关联和较大分子体系时,通常面临计算资源需求随体系规模急剧增长的挑战。近年来,变分量子本征求解器(VQE)被认为是近期含噪声中等规模量子设备上开展量子化学计算的重要候选方案。然而,将VQE直接用于分子几何优化时,仍存在两方面瓶颈:一是所需量子比特数随分子轨道数迅速增加;二是传统“外层几何优化—内层量子能量求解”的嵌套迭代流程计算代价高、收敛效率低,难以推广到更具化学实际意义的大分子体系。
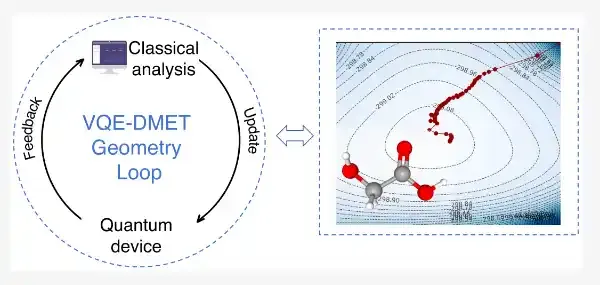
针对上述难题,研究团队提出了一种融合密度矩阵嵌入理论(DMET)与变分量子本征求解器(VQE)的量子—经典混合协同优化框架。该方法利用DMET将大分子体系划分为若干较小片段,在保持关键电子关联信息的同时显著降低量子计算所需资源;同时,团队将分子几何参数、VQE变分参数以及嵌入势的优化纳入统一流程,避免了传统方法中反复调用完整量子能量求解的高开销嵌套循环。
与以往先固定分子构型、再求解电子基态能量、最后更新几何参数的流程不同,该研究提出的VQE-DMET协同优化策略能够在同一迭代框架下同步推进电子结构优化与分子构型优化。这一设计不仅提升了收敛效率,也为在近期小规模量子设备上开展更大分子的几何优化提供了可行路径。
研究团队首先在H₄和H₂O₂等基准分子体系上验证了该方法的准确性与稳定性。结果表明,该框架能够得到与高精度经典参考方法一致的平衡几何结构,同时显著减少量子资源消耗。例如,在H₄体系中,DMET嵌入策略将所需量子比特数由8个降低至4个;在H₂O₂体系中,所需量子比特数由24个降低至16个,并成功获得接近参考值的结构参数。
在此基础上,研究团队进一步将方法应用于乙醇酸(C₂H₄O₃)分子的几何优化。乙醇酸是一类具有代表性的α-羟基酸分子,其体系规模和结构复杂度明显高于以往量子几何优化研究中常见的小分子模型。研究中,团队重点考察了羟基相关转动自由度的优化问题,并将原本需要58个量子比特描述的体系压缩至20个量子比特。计算结果显示,该方法能够有效收敛至与经典CCSD参考势能面一致的低能区域,展示了量子—经典协同优化方法在更复杂化学体系中的应用潜力。
该研究表明,通过将量子嵌入思想与变分量子算法深度结合,可以在保证化学精度的同时显著降低量子计算资源需求,为突破近期量子设备在分子模拟中的规模限制提供了新思路。该框架有望进一步拓展至复杂催化体系、药物分子、功能材料以及周期性体系的结构预测与性质计算,为面向真实化学问题的量子计算应用奠定基础。