深研院新材料学院潘锋团队运用多尺度拓扑和AI提升“物质多体相互作用”能量预测精度
在材料科学领域,准确理解多原子系统的行为是一项基础而又充满挑战的任务。以在锂离子等高能量密度电池中发挥关键作用的锂元素为例,精确预测锂原子簇内部的能量和相互作用,对于推动下一代储能技术至关重要。然而,随着原子数量的增加,系统交互的复杂性呈指数级增长。虽然深度学习模型潜力巨大,但在材料科学领域,高质量数据的稀缺性和模型的“黑箱”特性限制了其应用。
北京大学深圳研究生院新材料学院潘锋教授团队长期致力于图论结构化学方法的拓展与应用,作为一种将材料微观结构映射为数学图论和拓扑模型的方法,图论结构化学近年来在材料基因组、催化活性探索等方向中表现出强大的表示和预测能力,并在该领域提出了一系列创新性工作,实现了材料结构表示(J. Phys. Chem. Lett., 2023, 14: 954)、材料的反向设计(npj Comput. Mater., 2025, 11: 147)、新型固态电解质的设计(J. Am. Chem. Soc. 2024, 146, 18535;2025, DOI: 10.1021/jacs.5c04828 )、化学反应路径搜索(CCS Chemistry 2024, 7, 1)、催化活性相搜索(Nat Comm. 2025,16, 2542)。近日,潘锋团队与密歇根州立大学魏国卫(Guo-Wei Wei)教授团队合作,提出了一种名为“多尺度拓扑学习(Multiscale Topological Learning, MTL)”的可解释性框架,旨在提高多原子体系能量预测的准确性。该研究利用一种名为“持续拓扑拉普拉斯(Persistent Topological Laplacians, PTLs)”的多尺度拓扑方法,有效捕捉多体相互作用的内在属性。相关研究成果以“Enhancing Energy Predictions in Multi-Atom Systems with Multiscale Topological Learning”为题,发表于国际材料与化学的知名学术期刊《材料化学杂志A》( Journal of Materials Chemistry A)(2025,DOI: 10.1039/d5ta02687c)。
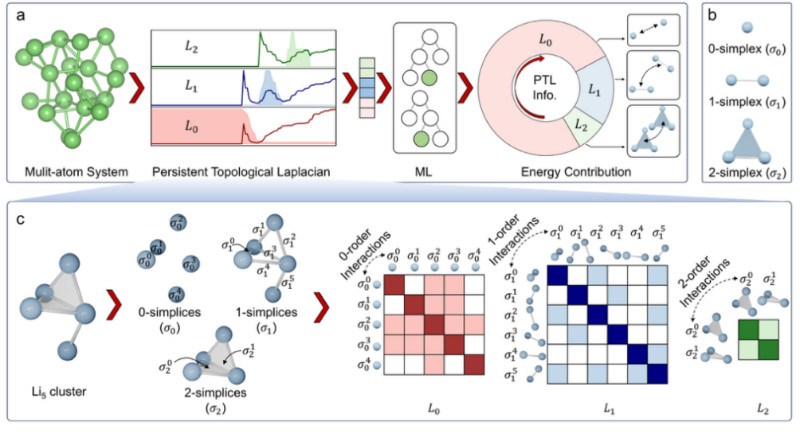
该研究的核心思想是将复杂的原子结构映射为拓扑空间中的单纯复形(simplicial complex),并运用PTL方法在多个尺度上进行分析,从而为原子结构生成能够表征其多体相互作用的“拓扑指纹”。其创新工作流程如下(图1):首先,将一个多原子系统(如锂原子簇)视为一个单纯复形,其中0维单形代表原子,1维单形代表原子间的连接,2维单形代表由三个原子组成的环状结构。接着,通过一个名为“过滤(filtration)”的参数(原子间距d)从小到大变化,该框架可以捕捉不同尺度下的结构。在每个尺度下,PTL方法会计算出0维、1维和2维的拓扑拉普拉斯算子(L0,L1,L2),这些算子的谱信息构成了对系统拓扑和几何特性的多维度描述。这种方法的新颖之处在于,它超越了传统的基于原子坐标的描述符,能够系统性地编码原子间“多体相互作用”的复杂信息,为理解和预测材料性质提供了全新的物理视角。
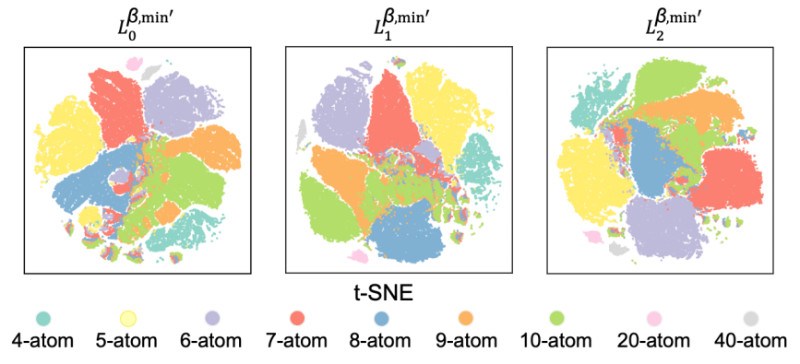
研究团队利用一个包含136,287个锂原子簇(涵盖4原子到40原子等多种体系)的大型数据。研究的主要结论是,引入更高维度的拓扑信息能持续提升能量预测的精度。具体而言,在预测任务中,随着1维(β1)和2维(β2)拓扑特征的加入,模型的平均绝对误差(MAE)显著降低。对于所有原子体系,包含0、1、2维信息的特征集(β012)均比只含0维信息(β0)或0、1维信息(β01)的特征集表现更优。
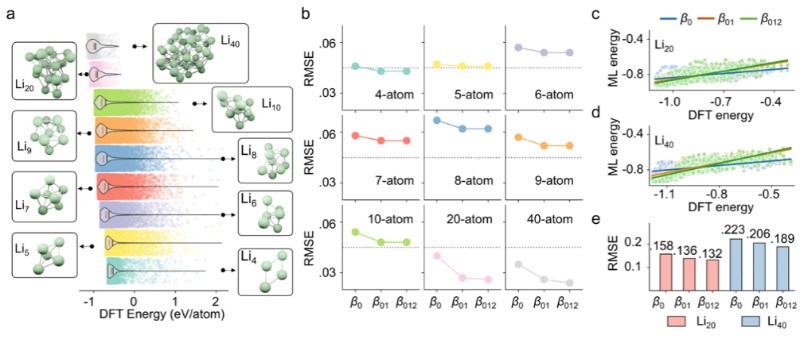
然而,尽管高维信息至关重要,但其对预测精度的贡献度随维度增加而逐渐减小。0维特征贡献最大,其次是1维特征。这一趋势与多体物理学理论高度一致,即高阶相互作用通常被视为对系统的微扰修正,从而验证了该拓扑方法的物理可解释性。此外,该框架还展现出强大的泛化与排序能。当研究人员使用小尺寸原子簇(Li4-Li10)训练模型后,该模型能有效预测从未见过的、结构更复杂的大尺寸原子簇(Li20-Li40)。结果显示,引入高维信息后,模型对Li20和Li40能量排序的准确性(通过皮尔逊相关系数PCC衡量)得到了大幅提升,这对从庞大的结构空间中筛选出最稳定的构型具有重要意义。
这项研究表明,基于拓扑学的机器学习框架不仅能精确预测多原子体系的能量,还能提供符合物理直觉的深刻见解。该方法有望被推广至更广泛的领域,如催化剂设计、新材料发现以及药物分子设计,为解决复杂体系中的科学难题开辟了新的道路。
潘锋与魏国卫为本文的通讯作者。深研院新材料学院博士毕业生陈冬为本文第一作者(现在在密歇根州立大学任助理研究员)。本研究由国家自然科学基金、广东省重点实验室等项目资助。








