中国科大实现分子—金属表面振动传能动力学的第一性原理精准计算
近日,中国科学技术大学蒋彬教授课题组在分子—金属表面振动传能模拟方面取得重要进展。研究成果以“分子从金属表面散射过程中振动能量转移的具有第一性原理精度的全维动力学模拟”(First-principles full-dimensional modelling ofvibrational energy transfer of moleculescattering from metal surfaces)”为题,11月26日发表在《自然·通讯》上,并被选为“编辑推荐”文章。
分子与金属表相互作用时,分子振动能量的转移在其中起着重要的作用。由于金属具有连续的电子能级结构,分子振动不仅可以通过传统的绝热途径与分子平动、转动以及表面声子发生耦合,还可以通过激发金属中的电子—空穴对发生非绝热能量转移。大量研究表明,这种振动—电子耦合介导的能量转移广泛存在于各种物理化学过程中,是决定分子振动寿命、金属电子发射以及等离激元催化等关键过程的重要机制。其中激发态NO分子在Au(111)表面的散射过程,被作为研究表面振动能量转移的代表性模型体系,在近25年被实验和理论广泛研究。该体系积累了大量态到态散射动力学的实验数据,成为验证金属表面能量转移动力学理论研究最好的试验场。此外,其中分子振动与平动是否通过电子耦合以及立体动力学的动力学机制等问题仍困扰着学界,尚未得到定量理论解释。
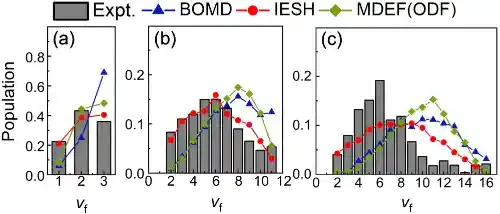
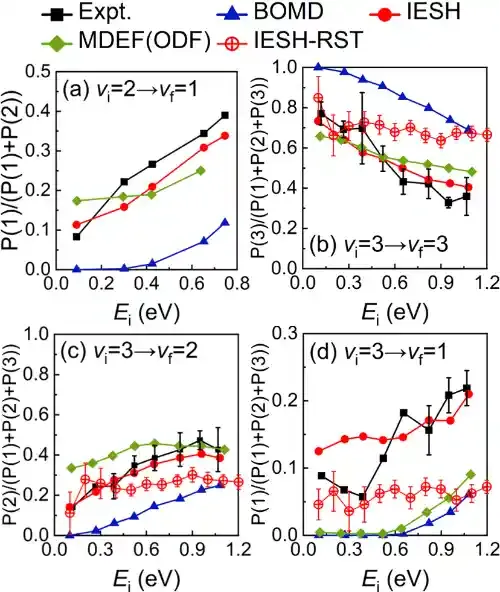
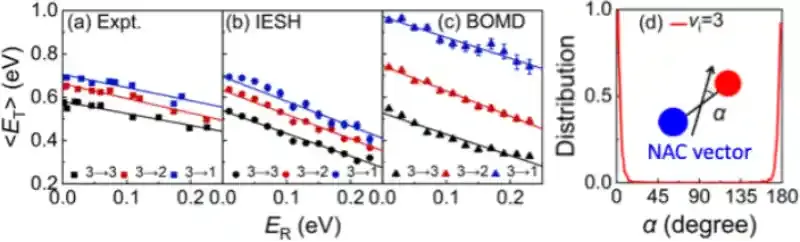
在此背景下,研究团队结合约束密度泛函理论、神经网络势能面和独立电子面跳跃(IESH)算法,发展了第一性原理精度的全维非绝热动力学模拟方法,系统性研究了NO分子从Au(111)表面散射过程中的能量转移动力学。新的计算结果在很大范围内重复实验结果,精度远超以前基于经验势函数和基于平均场近似的电子摩擦理论的计算结果(图1)。在分子—金属散射过程中,分子振动能量既可以通过非绝热途径转移到金属电子自由度,也可以通过绝热机制直接耦合到分子平动自由度(图2);而分子平动与金属电子之间的直接耦合则可以忽略不计,这些结果为不同自由度之间的能量传递提供了清晰的理论图像。
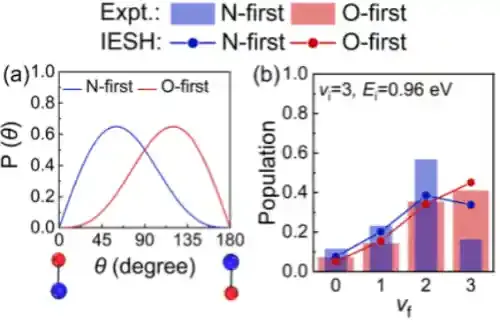
模拟结果还显示,在不同的初始振动态下,分子振动弛豫过程对分子取向的依赖性会发生变化,与实验测量吻合(图3)。进一步分析表明,该取向效应源于金属向分子电子转移过程的能垒对分子取向的依赖。
这套模拟策略不依赖经验参数,因而具备很强的普适性。它不仅适用于分子散射问题,也为研究更复杂的界面过程,比如光/电化学过程和等离激元催化,提供了一套可行的模拟方法。
中国科学技术大学化学物理系博士后孟刚为该论文的第一作者,蒋彬为通讯作者。该工作得到了量子科学与技术创新项目、中国科学院战略先导科技专项、攻坚专项、基金委杰出青年基金、创新群体等基金的资助。








